La ELA afecta aproximadamente a 2-3 personas por cada 100.000 habitantes. La mayoría de los casos son esporádicos (90-95%), mientras que un 5-10% son hereditarios. La enfermedad suele manifestarse entre los 40 y 70 años, siendo ligeramente más frecuente en hombres.
Síntomas
- Debilidad muscular: Comienza típicamente en manos, pies o extremidades y progresa
- Fasciculaciones: Contracciones musculares involuntarias visibles bajo la piel
- Calambres musculares: Especialmente nocturnos
- Dificultad para hablar (disartria): Habla arrastrada o nasal
- Dificultad para tragar (disfagia): Atragantamientos frecuentes
- Insuficiencia respiratoria: Por debilidad de los músculos respiratorios
Característicamente, la ELA respeta las funciones cognitivas, la sensibilidad, el control de esfínteres y los movimientos oculares hasta fases muy avanzadas.
Diagnóstico
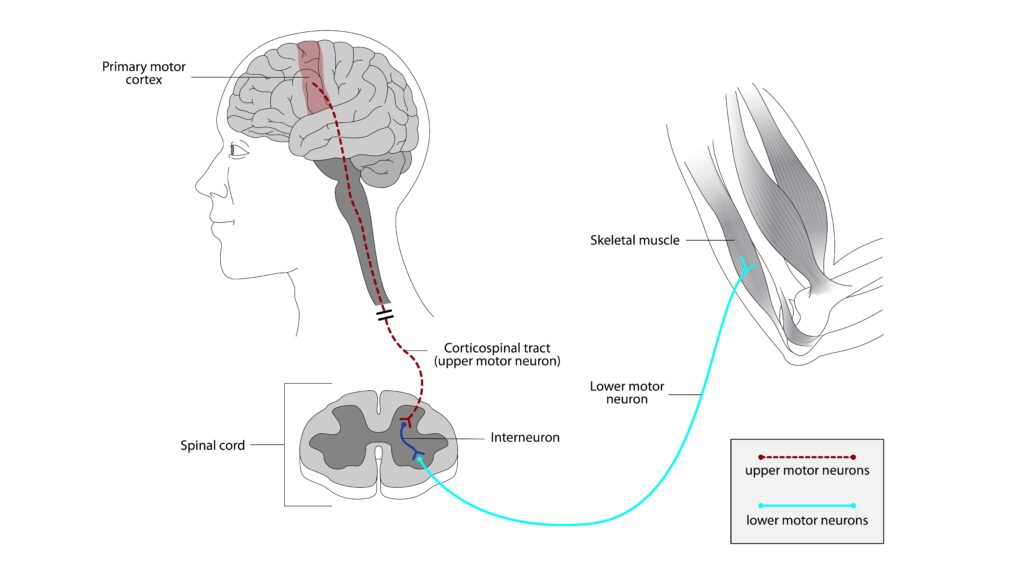
No existe una prueba única para diagnosticar la ELA. El diagnóstico se basa en los criterios de El Escorial, que requieren evidencia de degeneración de neurona motora superior e inferior, progresión de los síntomas y exclusión de otras enfermedades. La electromiografía (EMG) y los estudios de conducción nerviosa son fundamentales.
Tratamiento
No existe cura para la ELA. El riluzol, el primer fármaco aprobado, prolonga modestamente la supervivencia (2-3 meses). El tratamiento se centra en el manejo sintomático: fisioterapia, logopedia, soporte nutricional (gastrostomía), ventilación no invasiva y cuidados paliativos multidisciplinares.
Pronóstico
La supervivencia media desde el diagnóstico es de 2-5 años, aunque aproximadamente un 10% de los pacientes sobreviven más de 10 años. La causa de muerte suele ser la insuficiencia respiratoria. El físico Stephen Hawking, diagnosticado a los 21 años, sobrevivió 55 años con la enfermedad, un caso excepcional.
Preguntas frecuentes
La ELA clásica no afecta las funciones cognitivas. Los pacientes mantienen su inteligencia, memoria y capacidad de pensamiento. Sin embargo, un 15-20% puede desarrollar demencia frontotemporal asociada.
Solo el 5-10% de los casos son hereditarios (ELA familiar). El 90-95% son esporádicos, sin causa genética identificable. Se han identificado más de 20 genes asociados a la forma familiar.
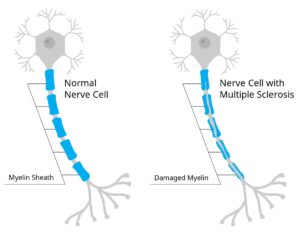
Son enfermedades diferentes. La ELA afecta las neuronas motoras causando debilidad y parálisis progresiva. La esclerosis múltiple es una enfermedad autoinmune que afecta la mielina causando síntomas variados (visuales, sensitivos, motores) con brotes y remisiones.
La esperanza de vida media tras el diagnóstico es de 2-5 años, aunque existe una gran variabilidad. El 10% de los pacientes sobrevive más de 10 años. La forma de inicio (bulbar frente a espinal), la edad al diagnóstico y la velocidad de progresión son los principales factores pronósticos.
Actualmente no hay cura para la ELA. El riluzol fue el primer fármaco aprobado y retrasa la progresión modestamente (unos 2-3 meses). El edaravone y el tofersen (para formas con mutación SOD1) son más recientes. El tratamiento es principalmente de soporte: ventilación no invasiva, nutrición, fisioterapia y cuidados paliativos.
Aunque clásicamente se considera una enfermedad solo motora, entre el 30-50% de los pacientes presentan algún grado de deterioro cognitivo frontotemporal y el 5-15% desarrollan demencia frontotemporal (DFT) completa. La evaluación neuropsicológica forma parte del seguimiento integral del paciente.
La ventilación no invasiva con mascarilla (VNI) es la intervención que más prolonga la supervivencia y mejora la calidad de vida en la ELA, en promedio 7-12 meses. Se inicia cuando la capacidad vital desciende por debajo del 50% o aparecen síntomas de insuficiencia respiratoria nocturna.
El 90% de los casos son esporádicos (sin antecedentes familiares). El 10% restante es familiar, con mutaciones en genes como SOD1, C9orf72, FUS o TARDBP. La mutación C9orf72 es la más frecuente tanto en formas familiares como esporádicas, presente en el 5-10% de todos los casos de ELA.